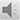

上次跟大家提到在我们开展RCT研究前,或者研究开始早期可以把临床试验方案以论文的形式向学术界汇报,这么做有助于提高研究的透明度,也有助于最终RCT研究结果的发表。可是,一般来说,RCT的设计方案或者说研究者手册内容非常多,我们应该把哪些内容摘出来作为公开发表的临床试验方案论文里的内容呢?今天就向大家介绍SPIRIT 2013声明,能帮助我们很好的形成一篇RCT的设计方案的论文。
SPIRIT 2013声明全称是:Standard Protocol Items:Recommendations for Interventional Trial 2013,顾名思义是适用于干预试验标准方案的推荐条目。其中包括了33个条目。SPIRIT 2013是2007年启动的SPIRIT国际合作项目,是广泛咨询了115为临床研究利益相关方专家(包括30名研究者,31名医疗专业人员,34名方法学家16名统计学家,14名试验协调者,15名期刊编辑,17名伦理委员会代表,7名企业和非企业的试验资助方,7名监管机构成员)后形成的。
根据SPIRIT2013声明的定义,研究方案是一份提供了足够细节的文件,旨在让人民理解研究项目的研究背景、理念、目的、研究人群、干预措施、方法、统计分析、伦理考虑、传播计划、研究的形成管理等;同时了解研究方法和实施中关键方面的可重复性;以及为伦理学批准到试验结果传播过程中对试验科学性和伦理学严谨性的评价提供依据。
从目前SPIRIT 2013声明的条目看来,SPIRIT 2013声明应该最适用于随机对照试验方案,但是,对那些非随机的干预性研究也同样可以参考SPIRIT 2013声明的条目选择合适的条目对研究设计方案进行阐述,方便读者了解研究的整体设计。下面是SPIRIT 2013声明的具体内容,共大家参考。中文的条目内容可以查阅本文的参考文献2。
SPIRIT 2013Checklist: Recommended Items to Address in a Clinical Trial Protocol andRelated Documents*
|
Section/Item |
Item No. |
Description |
|
Administrative information |
||
|
Title |
1 |
Descriptive title identifying the study design, population, interventions, and, if applicable, trial acronym |
|
Trial registration |
2a |
Trial identifier and registry name. If not yet registered, name of intended registry. |
|
2b |
All items from the World Health Organization Trial Registration Data Set (Appendix Table, available at www.annals.org) |
|
|
Protocol version |
3 |
Date and version identifier |
|
Funding |
4 |
Sources and types of financial, material, and other support |
|
Roles and responsibili-ties |
5a |
Names, affiliations, and roles of protocol contributors |
|
5b |
Name and contact information for the trial sponsor |
|
|
5c |
Role of study sponsor and funders, if any, in study design; collection, management, analysis, and interpretation of data; writing of the report; and the decision to submit the report for publication, including whether they will have ultimate authority over any of these activities |
|
|
5d |
Composition, roles, and responsibilities of the coordinating center, steering committee, end point adjudication committee, data management team, and other individuals or groups overseeing the trial, if applicable (see item 21a for DMC) |
|
|
Introduction |
||
|
Background and rationale |
6a |
Description of research question and justification for undertaking the trial, including summary of relevant studies (published and unpublished) examining benefits and harms for each intervention |
|
6b |
Explanation for choice of comparators |
|
|
Objectives |
7 |
Specific objectives or hypotheses |
|
Trial design |
8 |
Description of trial design, including type of trial (e.g., parallel group, crossover, factorial, single group), allocation ratio, and framework (e.g., superiority, equivalence, noninferiority, exploratory) |
|
Methods Participants, interventions, and outcomes |
||
|
Study setting |
9 |
Description of study settings (e.g., community clinic, academic hospital) and list of countries where data will be collected. Reference to where list of study sites can be obtained |
|
Eligibility criteria |
10 |
Inclusion and exclusion criteria for participants. If applicable, eligibility criteria for study centers and individuals who will perform the interventions (e.g., surgeons, psychotherapists) |
|
Interventions |
11a |
Interventions for each group with sufficient detail to allow replication, including how and when they will be administered |
|
11b |
Criteria for discontinuing or modifying allocated interventions for a given trial participant (e.g., drug dose change in response to harms, participant request, or improving/worsening disease) |
|
|
11c |
Strategies to improve adherence to intervention protocols, and any procedures for monitoring adherence (e.g., drug tablet return, laboratory tests) |
|
|
11d |
Relevant concomitant care and interventions that are permitted or prohibited during the trial |
|
|
Outcomes |
12 |
Primary, secondary, and other outcomes, including the specific measurement variable (e.g., systolic blood pressure), analysis metric (e.g., change from baseline, final value, time to event), method of aggregation (e.g., median, proportion), and time point for each outcome. Explanation of the clinical relevance of chosen efficacy and harm outcomes is strongly recommended |
|
Participant timeline |
13 |
Time schedule of enrollment, interventions (including any runins and washouts), assessments, and visits for participants. A schematic diagram is highly recommended (Figure). |
|
Sample size |
14 |
Estimated number of participants needed to achieve study objectives and how it was determined, including clinical and statistical assumptions supporting any sample size calculations |
|
Recruitment |
15 |
Strategies for achieving adequate participant enrollment to reach target sample size |
|
Assignment of interventions (for controlled trials) |
||
|
Allocation Sequence generation |
16a |
Method of generating the allocation sequence (e.g., computer-generated random numbers), and list of any factors for stratification. To reduce predictability of a random sequence, details of any planned restriction (e.g., blocking) should be provided in a separate document that is unavailable to those who enroll participants or assign interventions. |
|
Allocation concealment mechanism |
16b |
Mechanism of implementing the allocation sequence (e.g., central telephone; sequentially numbered, opaque, sealed envelopes), describing any steps to conceal the sequence until interventions are assigned |
|
Implementation |
16c |
Who will generate the allocation sequence, who will enroll participants, and who will assign participants to interventions |
|
Blinding (masking) |
17a |
Who will be blinded after assignment to interventions (e.g., trial participants, care providers, outcome assessors, data analysts), and how |
|
Blinding (masking) |
17b |
If blinded, circumstances under which unblinding is permissible, and procedure for revealing a participant’s allocated intervention during the trial |
|
|
||
|
Data collection, management, and analysis |
||
|
Data collection methods |
18a |
Plans for assessment and collection of outcome, baseline, and other trial data, including any related processes to promote data quality (e.g., duplicate measurements, training of assessors) and a description of study instruments (e.g., questionnaires, laboratory tests) along with their reliability and validity, if known. Reference to where data collection forms can be found, if not in the protocol. |
|
18b |
Plans to promote participant retention and complete follow-up, including list of any outcome data to be collected for participants who discontinue or deviate from intervention protocols |
|
|
Data management |
19 |
Plans for data entry, coding, security, and storage, including any related processes to promote data quality (e.g., double data entry; range checks for data values). Reference to where details of data management procedures can be found, if not in the protocol. |
|
Statistical methods |
20a |
Statistical methods for analyzing primary and secondary outcomes. Reference to where other details of the statistical analysis plan can be found, if not in the protocol. |
|
20b |
Methods for any additional analyses (e.g., subgroup and adjusted analyses) |
|
|
Statistical methods |
20c |
Definition of analysis population relating to protocol nonadherence (e.g., as-randomized analysis), and any statistical methods to handle missing data (e.g., multiple imputation) |
|
Monitoring |
||
|
Data monitoring |
21a |
Composition of DMC; summary of its role and reporting structure; statement of whether it is independent from the sponsor and competing interests; and reference to where further details about its charter can be found, if not in the protocol. Alternatively, an explanation of why a DMC is not needed. |
|
21b |
Description of any interim analyses and stopping guidelines, including who will have access to these interim results and make the final decision to terminate the trial |
|
|
Harms |
22 |
Plans for collecting, assessing, reporting, and managing solicited and spontaneously reported adverse events and other unintended effects of trial interventions or trial conduct |
|
Auditing |
23 |
Frequency and procedures for auditing trial conduct, if any, and whether the process will be independent from investigators and the sponsor |
|
Ethics and dissemination |
||
|
Research ethics approval |
24 |
Plans for seeking REC/IRB approval |
|
Protocol amendments |
25 |
Plans for communicating important protocol modifications (e.g., changes to eligibility criteria, outcomes, analyses) to relevant parties (e.g., investigators, RECs/IRBs, trial participants, trial registries, journals, regulators) |
|
Consent or assent |
26a |
Who will obtain informed consent or assent from potential trial participants or authorized surrogates, and how (see item 32) |
|
26b |
Additional consent provisions for collection and use of participant data and biological specimens in ancillary studies, if applicable |
|
|
Confidentiality |
27 |
How personal information about potential and enrolled participants will be collected, shared, and maintained in order to protect confidentiality before, during, and after the trial |
|
Declaration of interests |
28 |
Financial and other competing interests for principal investigators for the overall trial and each study site |
|
Access to data |
29 |
Statement of who will have access to the final trial data set, and disclosure of contractual agreements that limit such access for investigators |
|
Ancillary & post-trial care |
30 |
Provisions, if any, for ancillary and post-trial care, and for compensation to those who suffer harm from trial participation |
|
Dissemination policy |
31a |
Plans for investigators and sponsor to communicate trial results to participants, health care professionals, the public, and other relevant groups (e.g., via publication, reporting in results databases, or other data-sharing arrangements), including any publication restrictions |
|
31b |
Authorship eligibility guidelines and any intended use of professional writers |
|
|
31c |
Plans, if any, for granting public access to the full protocol, participant-level data set, and statistical code |
|
|
Appendices |
||
|
Informed consent materials |
32 |
Model consent form and other related documentation given to participants and authorized surrogates |
|
Biological specimens |
33 |
Plans for collection, laboratory evaluation, and storage of biological specimens for genetic or molecular analysis in the current trial and for future use in ancillary studies, if applicable |
DMC = datamonitoring committee; IRB = institutional review board; REC = research ethicscommittee; SPIRIT = Standard Protocol Items: Recommendations for InterventionalTrials.
参考文献:
1. Chan AW, et.al.SPIRIT 2013 Statement: Defining Standard Protocol Items forClinical Trials. Ann Intern Med. 2013, 158(3):200-7.
2. Chan AW, et.al.SPIRIT 201 3声明:定义临床研究方案的标准条目. 中国循证医学杂志. 2013, 13(12):1501-1507